高效液相色谱检测水产品中甲氧苄啶残留量
2019-07-26 13:55:54 来源: 食品安全导刊
摘要:本文建立了水产品中甲氧苄啶残留量的高效液相色谱(HPLC)的检测方法。该方法操作简便,准确度高,适用于水产品中甲氧苄啶残留检测。
关键词:高效液相色谱;水产品;甲氧苄啶
目前,水产品中甲氧苄啶的检测方法主要有紫外分光光度法[4-5]、吸附溶出伏安法[6]、液相色谱法等[7-9]。本文以市售的鲜鱼为研究对象,对其中的甲氧苄啶检测前处理条件进行了选择和优化,高效液相色谱紫外检测器检测条件的建立和优化,建立了高效液相色谱法检测水产品中甲氧苄啶的方法。
结果与讨论
样品前处理条件的优化
(1)提取溶剂的选择。水产品基质复杂,且含有较多的脂肪和蛋白等成分,而甲氧苄啶属于弱碱性化合物,易溶于酸性溶液,而纯水相的提取液在净化时易出现乳化,不利于分层,因此本研究中选择酸化的甲醇作为提取液。考虑样品基质中含有蛋白,硫酸具有一定的沉淀蛋白的作用,因此选择硫酸化的甲醇溶液,分别考察了甲醇-硫酸体积比为7:3、6:4、5:5、4:6、3:7时对水产品中甲氧苄啶的提取效果,当比例为7:3时效果最好,因此最终选择甲醇-硫酸体积比7:3的溶液作为提取液。
(2)净化条件的优化。在酸性条件下,甲氧苄啶几乎不溶于三氯甲烷,而水产品中含有较多的脂肪、蛋白和部分色素等,三氯甲烷可以除去脂。分别考察了在提取过程中和提取之后加入三氯甲烷的净化和回收效果,发现在提取过程中加入三氯甲烷,首次提取液未见溶入三氯甲烷,但在第二次提取过程中,有部分三氯甲烷溶于提取液中,提取液体积明显大于理论加入体积,最终造成提取液中溶入了三氯甲烷相,引入杂质,净化效果变差,同时因杂质的引入,使得回收率不到70.0%;与此同时我们在提取过程之后向合并后的提取液中加入三氯甲烷,未发现有三氯甲烷相溶于提取液中的情况,且回收率均在80.0%以上,因此选择在提取过程之后加入三氯甲烷进行净化。
(3)线性方程与定量限。按照1.4项的方法配制6水平标准曲线,并在优化色谱条件下进行检测,以甲氧苄啶的峰面积为纵坐标,甲氧苄啶的浓度为横坐标绘制曲线,甲氧苄啶的线性范围为0.05~1.0 μg/mL,相关系数r为0.9998,线性关系良好,定量限为15 μg/kg。
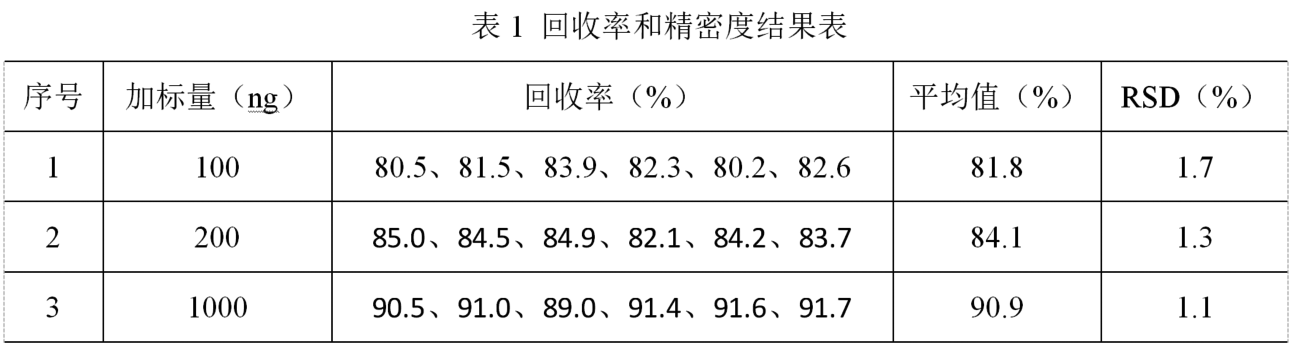
(4)回收率和精密度。在空白样品中添加甲氧苄啶标准溶液,制备含量分别为20、40和200 μg/kg的加标样品,按照1.4项处理方法及仪器测定条件进行测试,每个添加水平平行测定6份,结果见表1。各化合物的平均回收率为80.2~91.7%,相对标准偏差为1.1~1.7%,采用本方法检测水产品中的甲氧苄啶含量,准确度和精密度均较好。
实际样品检测
采用本研究建立的检测方法,对市售的10批次鲜鱼进行检测,其中3批检出甲氧苄啶,但检出值均小于农业部235号公告附录2:已批准的动物性食品中最高残留限量规定的限量要求50 g/kg范围内,可以放心食用。
结论
本研究采用酸化的甲醇提取水产品中甲氧苄啶,用三氯甲烷除脂等杂质,二氯甲烷反萃取的方法,结果准确可靠,精密度高,重现性好。与国家标准《GB 29702-2013水产品中甲氧苄啶残留量的测定》相比,操作简单、快速,除杂效果更优,适用于食品检验检测机构进行水产品中的甲氧苄啶的检测。
佟晓波 孙晓娟 张京华
辽宁省食品检验检测院






热点推荐
-

主要食品配料厂商携手支持可持续农业
-

别样肉客在华推出脆香酥炸植物基蟹饼,为新春佳节增添美食新选
-

ADM首度亮相FBIF2023,探索食品饮料的今天、明天和未来
-

专访婴儿水团体标准制定者:为何为婴儿饮用水制定更高标准?
-

使用梅特勒-托利多X光机的五大理由
-

广西发动全区3·15食品安全“你点我检 服务惠民生”问卷调查
-

河南全省食品安全监管工作会议在鹤壁召开
-

万里挑一的“进博TOP好物”,三养创新产品与食安承诺受赞誉
-

河南省餐饮食品安全“总监话总监”巡讲活动在洛阳启动
-

郑州市举办2024年“质量月”质量诚信进商超暨质量提升交流观摩会
-

仁和中方医药股份联合民生大药房送健康捐赠活动在郑州举行
-

三养密阳工厂深度揭秘:火鸡面的食品安全与品质卓越之道
-

郑州市金水区召开学校食堂承包经营企业 食品安全行政指导会
-

陕西省举办全省学校食堂食品安全管理及投诉处置现场会
-

山西省局举办质量提升行动助力汾酒专业镇高质量发展活动
-

郑州市金水区开展肉类产品质量安全专项整治
-

郑州市市场监管局开展网络餐饮服务食品安全行政指导工作
-

西藏山南市率先将“两个责任”进党校 推动食品安全社会共治
-

湖北:开展校园食品安全和“五一” 期间食品安全监管工作
-

河南省政府食品安全办召开2024年夏季食品安全形势会商会议
-

山西省局召开食品安全抽检承检机构任务部署会
-

南昌市开展春节前食品经营安全监督检查
-

河南省汝南县:“你点我检服务惠民生”护航“双节”食品安全
-

湖北省局推动共建“外卖小哥食堂” 让外卖骑手暖心又暖胃
-

焦作市校园食品安全排查整治专项行动动员部署会召开
-

汝南县市场监管局“四个抓手”提升集中用餐单位食品安全水平
-

山西省市场监管局食品案件查办指导中心正式挂牌运行
-

河南省汝南县市场监管局多措并举开展学校食堂食品安全评价
-

恩施州市场监管局服务第八届世界硒博会
-

开封市市场监管局2023年食品抽检工作质量提升推进会召开